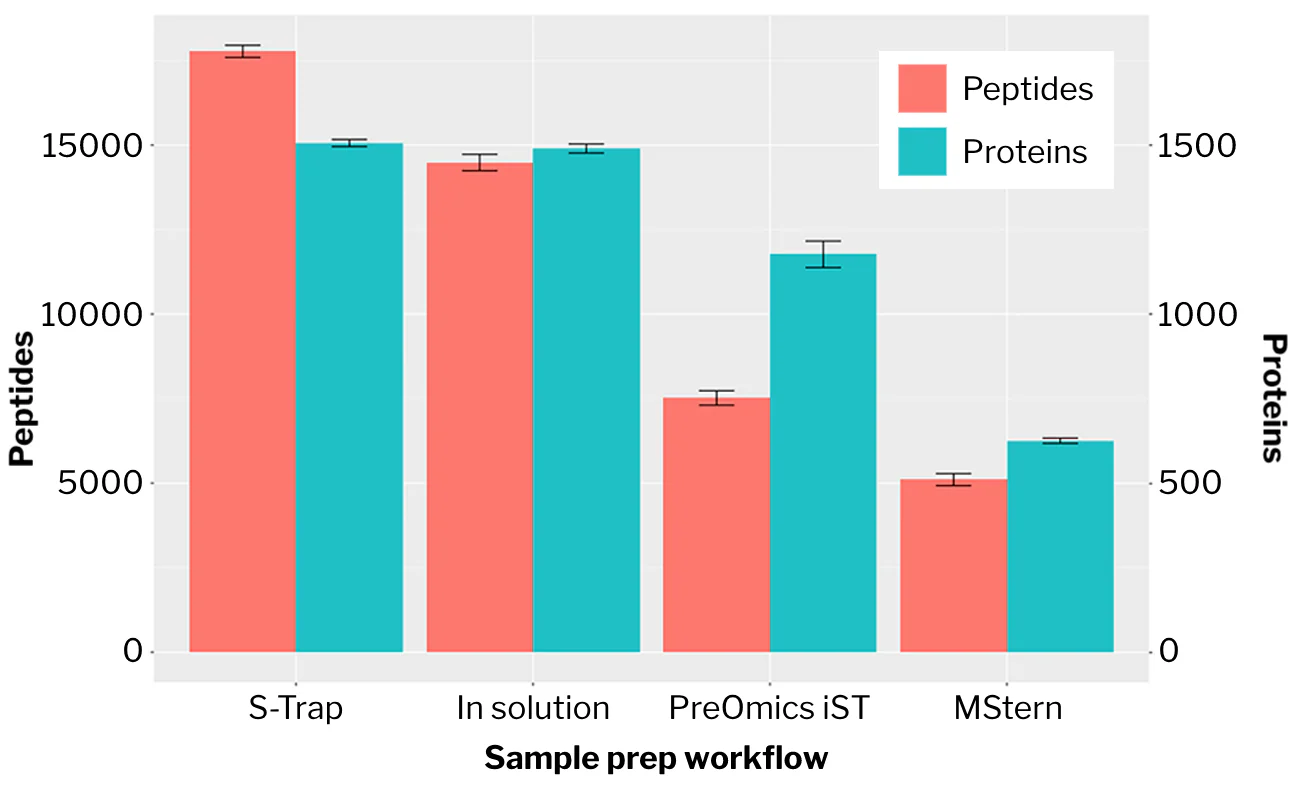
“[T]he S-Trap workflow gives the greatest number of peptide and protein identifications. Using the S-Trap method and starting with ∼0.5 mL [of urine], we identify ∼1500 protein groups and ∼17 700 peptides from DDA analysis with a single injection. … To our knowledge, these are the largest reported values for peptide and protein identification from urine samples using single-shot bottom-up proteomics.”
Publication: Ding, H., Fazelinia, H., Spruce, L. A., Weiss, D. A., Zderic, S. A., & Seeholzer, S. H. (2020). Urine proteomics: Evaluation of different sample preparation workflows for quantitative, reproducible and improved depth of analysis. Journal of Proteome Research. doi:10.1021/acs.jproteome.9b00772.
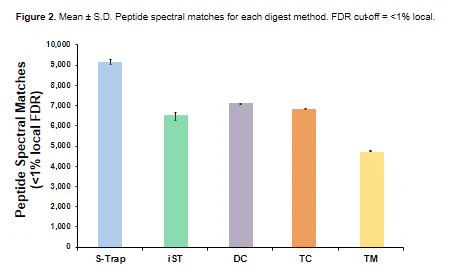
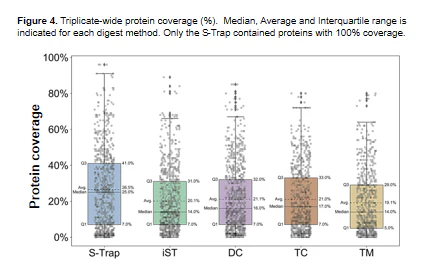
“S-Trap had the highest peptide recovery, highest number of peptide spectral matches (PSMs), and highest median coverage”
Poster: Neely BA, Bland A, Janech MG. Comparison of in-solution, FASP, and S-trap based digestion methods for bottom-up proteomic studies. NIST poster presented at ASMS 2019.
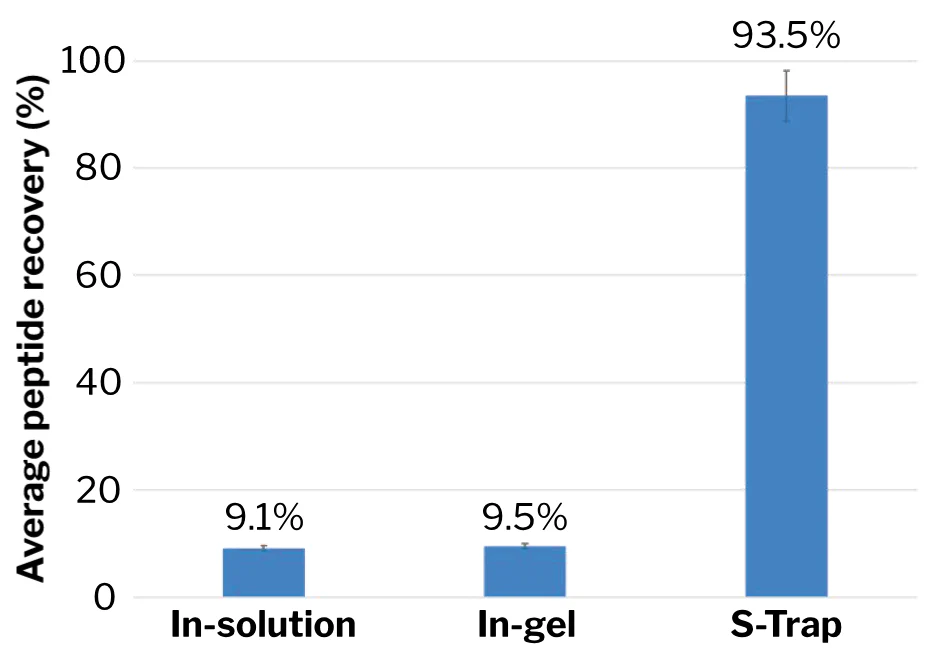
Proteomics analysis of clinical FFPE samples of primary colorectal adenocarcinoma. “[T]he average peptide recovery obtained by in-solution digestion, in-gel digestion and S-Trap digestion are 9.1%, 9.5% and 93.5%, respectively. … The conclusion of this study is basically the same as that of Katelyn [Ludwig] et al.” (Study immediately below.) 6,052 proteins were subsequently detected via S-Trap analysis of 58 colon cancer FFPE samples from the biobank of Memorial Sloan Kettering Cancer Center (MSKCC).
Dissertation: Study of proteomics on colon cancer FFPE tissue based on liquid chromatography-mass spectrometry. Zhou Yihua. Dissertation, May 2019, The Second Affiliated Hospital of Nanchang University. Fig. 3.1 (p. 21) and table 3.2 (p. 22), proportionately regraphed.
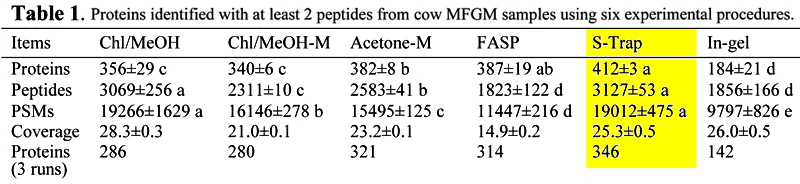
S-Traps yielded the highest reproducibility, highest number of PSMs and proteins/peptides identified in milk fat globule membrane (MFGM).
Publication: Yang Y, Anderson E, Zhang S. Evaluation of six sample preparation procedures for qualitative and quantitative proteomics analysis of milk fat globule membrane. Electrophoresis. 2018 Sep;39(18):2332-9.
“S-Traps demonstrated the best overall performance, with the largest numbers of protein identifications and quantitative reproducibility. [You can] use SDS in your proteomics sample [and] it outperformed all other methods regardless of lysis conditions. Therefore, S-Traps provide the best balance of time, cost, and performance of the bottom-up workflows examined in this study.”
Publication: Ludwig KR, Schroll MM, Hummon AB. Comparison of in-solution, FASP, and S-trap based digestion methods for bottom-up proteomic studies. Journal of proteome research. 2018 May 13;17(7):2480-90.
S-Traps “…can generate digestion-ready samples within minutes. As comparing to currently available approach such as FASP, the S-Trap truly shortens the bench time from over three hours to 10~20 minutes, thus speeding up the entire assembly line of proteomics analysis.”
Publication: HaileMariam M, Eguez RV, Singh H, Bekele S, Ameni G, Pieper R, Yu Y. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. Journal of proteome research. 2018 Aug 16;17(9):2917-24.
“We [evaluate] lysis buffers with S-Trap: SDS, urea, NP-40, RIPA, and SDS with DTT (SDT). We show that S-Trap is compatible with all of the tested buffers… [W]e anticipate that the method will transform experimental planning for mass-spectrometry-based proteomics, making it far more flexible and tolerable of various lysis buffers.”
Publication: Elinger D, Gabashvili A, Levin Y. Suspension trapping (S-Trap) is compatible with typical protein extraction buffers and detergents for bottom-up proteomics. Journal of proteome research. 2019 Feb 14;18(3):1441-5.
“Incubating mammalian cells in bioreactors requires the addition of polymeric surfactants such as Pluronic F68 [which] are incompatible with mass spectrometry proteomics and must be eliminated during sample preparation. S-Trap substantially reduced or eliminated the polymer(s) and S-Trap provided the most robust cleanup and highest quality data.”