One Workflow, Many Omics: Si-Trap™ Delivers Proteomic Depth and Metabolite Coverage

Related Articles



In collaboration with the Tamir Lab at UNC Chapel Hill, ProtiFi evaluated the Si-Trap™ as a true multi-omics workflow: one sample in, multiple omics out, without sacrificing analytical quality. Using MCF7 ER+ breast cancer cells, the Si-Trap™ was compared directly with a conventional urea-based proteomics method and a standard metabolite extraction. Si-Trap™ delivered greater proteome depth than urea while maintaining comparable metabolite detection.
Across every proteomic comparison, the Si-Trap™ identified more proteins than the urea method. This improvement was observed in global proteome profiling as well as phosphoproteome and tyrosine phosphoproteome analyses, showing that the benefit was not limited to a single protein readout (Figures 1 and 2). Rather than simplifying sample preparation at the expense of sensitivity, the Si-Trap™ increased protein identifications while still supporting metabolomics from the same preparation.
Metabolomics results were similarly encouraging. In positive ion mode, the Si-Trap™ matched the standard workflow, with 81 metabolites detected in each case. In negative ion mode, the Si-Trap™ detected 83 metabolites compared with 85 for the standard method. These findings show that Si-Trap™ preserved broad metabolite detection while delivering stronger proteomic depth (Figure 3).
The Si-Trap™ thus removes one of the biggest barriers in multi-omics research: the need to choose between simplicity and analytical depth. By generating high-quality metabolomic and proteomic fractions from a single workflow, Si-Trap™ helps researchers gain more insight from every sample.
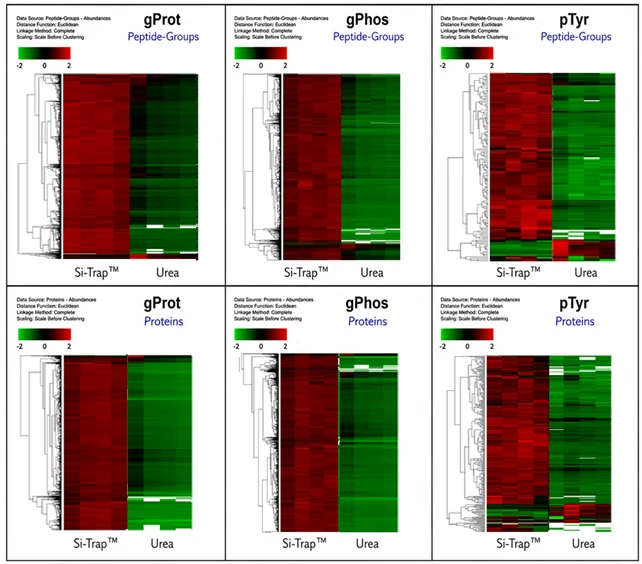
Figure 1: The Si-Trap™ Provides Better Proteome Coverage and Intensity Than Urea
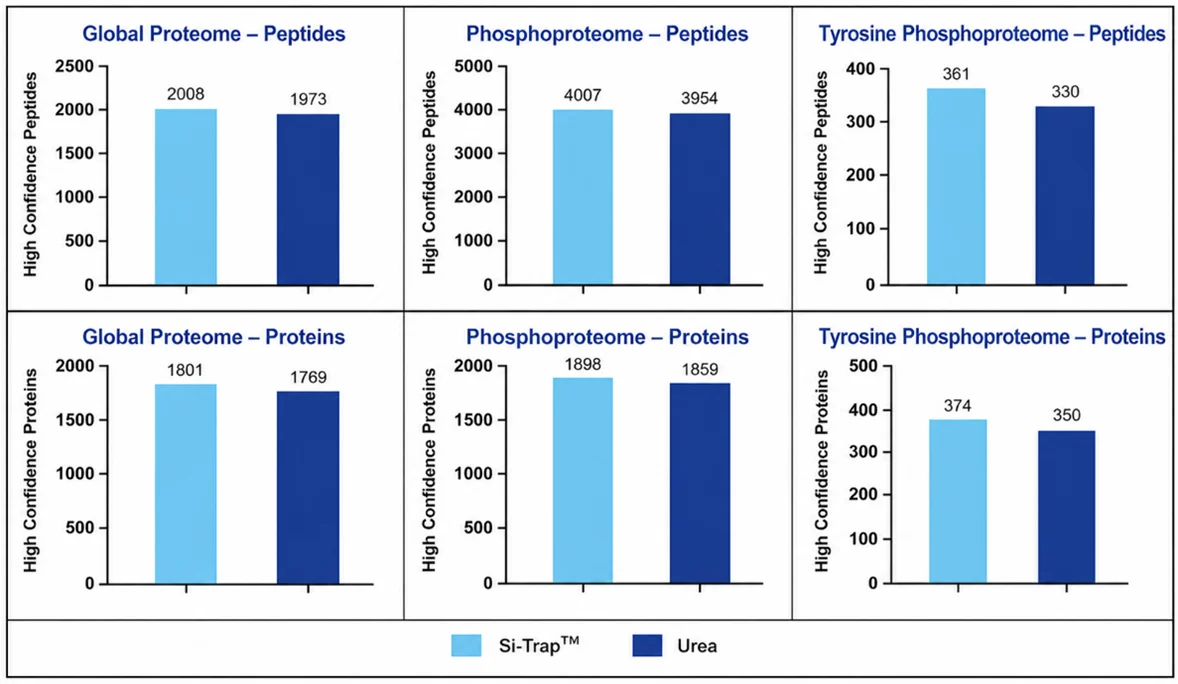
Figure 2: The Si-Trap™ demonstrates stronger Proteomic Depth Across Global and Phosphorylation Analyses
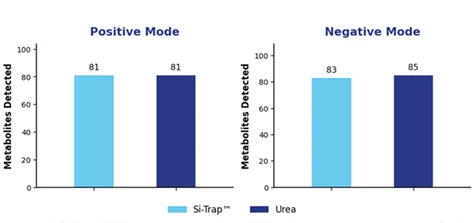
Figure 3: The Si-Trap™ Preserves Broad Metabolite Coverage