“…I was planning to spend all day analyzing a viral TMT PAWS dataset in R, but I uploaded it to simplifi simplifi.protifi.com and it did the comparisons I needed in minutes. with GO and pathways too! Now I need to find something else to do…”
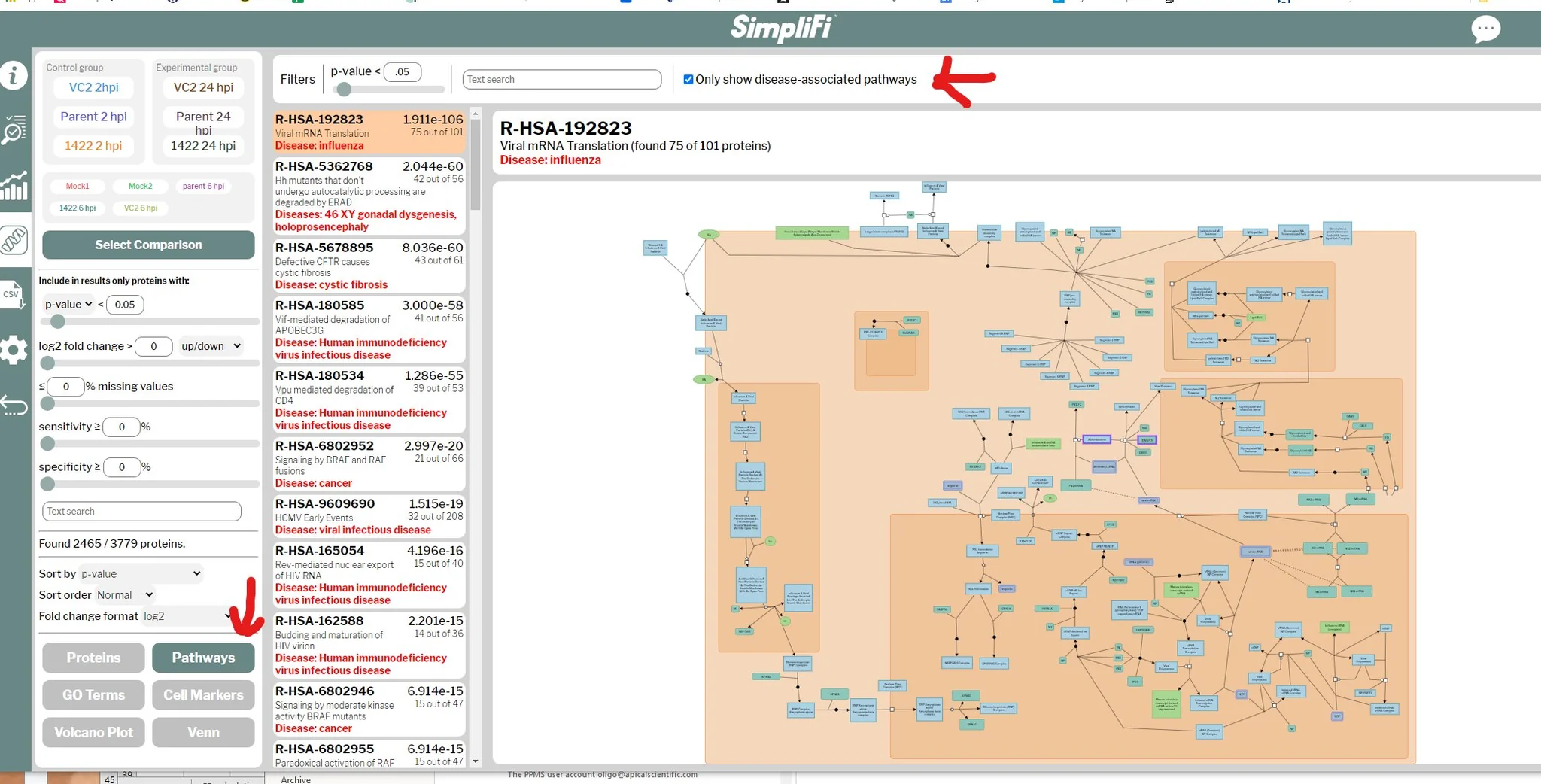
“My intern helped to do a quick & dirty comparison of few shotgun #proteomics sample preparation methods to figure out which one we should carry to next stage of automation… I think the answer is quite clear :-) @ProtifiLlc“
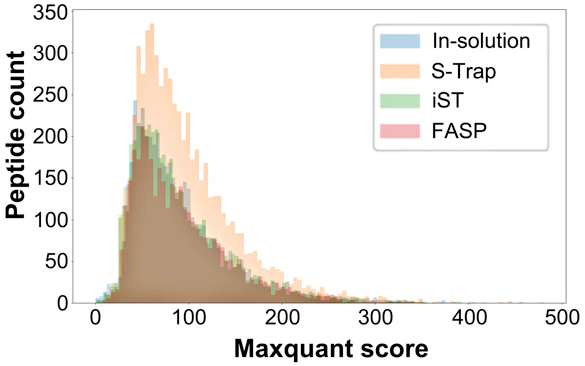
“Special thanks to @ProtifiLlc! S-Trap simply enables us to process hundreds of proteomics samples within one day, which guaranteed everything still ran smoothly even though I only work in lab one day per week.”
“[W]e found that the S-Traps demonstrated the best overall performance, with the largest numbers of protein identifications and quantitative reproducibility. The S-Trap protocol requires little more time than a typical in-solution digestion and provides the flexibility to use SDS in your proteomics sample. In addition, it outperformed all other methods regardless of lysis conditions. Therefore, S-Traps provide the best balance of time, cost, and performance of the bottom-up workflows examined in this study.“
Publication: Katelyn Ludwig, et al. Comparison of in-solution, FASP, and S-trap based digestion methods for bottom-up proteomic studies. Journal of proteome research. 2018 May 13;17(7):2480-90.
“[S-Traps] …can generate digestion-ready samples within minutes. As comparing to currently available approach such as FASP, the S-Trap truly shortens the bench time from over three hours to 10~20 minutes, thus speeding up the entire assembly line of proteomics analysis.”
Publication: Milkessa HaileMariam, et al. (2018) HaileMariam M, Eguez RV, Singh H, Bekele S, Ameni G, Pieper R, Yu Y. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. Journal of proteome research. 2018 Aug 16;17(9):2917-24.
“We [evaluate] lysis buffers with S-Trap: SDS, urea, NP-40, RIPA, and SDS with DTT (SDT). We show that S-Trap is compatible with all of the tested buffers… [W]e anticipate that the method will transform experimental planning for mass-spectrometry-based proteomics, making it far more flexible and tolerable of various lysis buffers.”
“1,245 proteins (min 2 peptides) from 500 clinical cells! Using @ProtifiLlc s-trap”
S-Traps yielded the highest reproducibility, highest number of PSMs and proteins/peptides identified in milk fat globule membrane (MFGM).
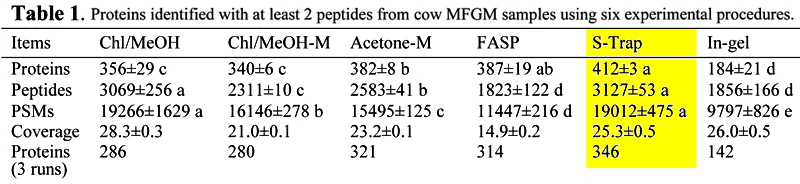
“The S-Trap has changed sample processing in our lab dramatically. Using SDS for sample preparation is no longer hampered by the need to precipitate and resolubilize sample pellets prior to digestion. The fast digestion times enhance our phosphopeptide recovery and enable a wide range of sample quantities (from 10 ug to 10 mg) to be processed efficiently.”
“I am very excited about ProtiFi S-Traps. We compared our standard preparation to S-Traps on clinical samples. The number of peptides identified increased 51% – 125% and proteins increased 29% – 54%. Simultaneously, the coefficient of variation (CV), decreased from 12% – 16% with standard methods to 5% – 9% with S-Traps. We can now perform deeper dives into the proteome while acquiring more reproducible data.”
“S-Trap columns have significantly streamlined and improved our proteomics workflows and they quickly became one of our favorite tools in sample preparation. SDS + S-Trap gives superior performance over standard urea based in-solution digestions: better yields, more identifications, and greater reproducibility, all in less time. They have been particularly useful for difficult samples requiring efficient extraction with SDS (they’re far better than FASP), samples with interferences requiring cleanup (such as detergents), and procedures requiring high recoveries and yields (such as PTM enrichments). Having multiple formats geared for different sample loads is also a huge plus. Highly recommended!”
“Our lab was a FASP-based clinical proteomics lab. We prepared all the proteomics samples using FASP-filters; all our publications are FASP-orientated; all our grants are FASP-related. Now, we have switched to ProtiFi’s S-Trap platform! As you can see from our preliminary data, S-Trap produce similar number of miscleavage sites, similar number of proteins/peptide IDs and basically have similar performance to FASP. But S-Trap is >20 times faster than FASP not counting the digestion step!”
Prof. Dr. Yanbao Yu, J. Craig Venter Institute, Rockville, MD
“We were very surprised with S-Traps studying bile. We identified 225 proteins analyzing 20 ug of protein with standard techniques. We then analyzed 1 ug using your “ultra-high recovery” micro protocol. We identified 662 proteins: 3X more! The reproducibility of peptide retention times is also significantly improved.”
Sergio Ciordia Higuera, National Center for Biotechnology, Universidad Autónoma de Madrid, Madrid, Spain
“The S-Trap is honestly AMAZING! We just finished processing approximately 200,000 cells for a complex analysis and compared it to FASP on our QE Plus. We found a significant increase in overall quantifiable peptides. The results gave us higher confidence in our analysis and we plan on purchasing at least 3 more boxes right away! S-Traps are simply fantastic!”
“ProtiFi’s S-Trap is a revolutionary tool to increase reproducibility and quality of traditional membrane proteomic analysis. As a lipid biologist, the analysis of enzymes localized to membrane bound organelles frequently frustrated our attempts to identify regulatory components of bioactive lipids. The ability of S-Traps to handle 5% SDS has dramatically increased our recovery of membrane bound proteins. There is no need to solubilize the proteins once they have been loaded onto the S-Trap, therefore reproducibility is increased as we no longer fight to keep these hydrophobic proteins in solution. The S-Trap is a great addition to our toolbox that has an easy and quick protocol.”
“We have tested various proteomics sample preparation protocols before we came to S-Traps. Protein digestion by using S-Traps is easy and fast. In terms of both qualitative and quantitative measurement, S-Traps provide much more reproducible throughput and results as compared to traditional in-solution digestion protocol and FASP.”
“The S-Trap is by far the best way to digest samples. The benefits include reliable SDS solubilization, significantly reduced sample handling (no need to MeOH:chloroform precipitate and struggle solubilizing the pellet again) as well as fast digestion times and instrument friendly output with little further clean-up needed.”
Proteomics Core Manager
“ProtiFi’s S-Trap columns work great. They outperformed urea and FASP protocols. The yield is higher, digestion takes one hour (!!) and the whole protocol is much simpler than FASP.”
Core Lab Head
“I work in a Core Facility for Proteomics at a federal institute. Most applications are standard workflows like Global Expression Analysis, Interaction analysis or PTMs. Samples are often prepared by our collaborators, which are not Mass Spec experts and we recommended FASP or in Solution Digestion (Urea) for a long time. However sample quality was always an issue (low yield and SDS in FASP and PEG in in-Solution). So I tested almost all Sample Prep Methods I know of, like inStageTip or SP3, etc. Your S-Traps perform very well and are highly reproducible. I perform Digestion in S-Traps overnight in 50 mM TEAB (pH = 8.0 @ 37 °C) and load samples without desalting, which is also very nice to skip this step. We now give the S-Traps to all our collaborators.”
Researcher, Proteomics Core Facility
Researcher, Proteomics Core Facility
“By processing FFPE samples with “S-Trap sample processing, HYPERsol will be suitable for the automated, high-throughput analysis of clinical specimens. We thus anticipate that the HYPERsol workflow will enable novel discoveries from rich clinically annotated and histologically characterized FFPE biorepositories worldwide, thereby helping to usher in a new era of clinical proteomics.”
Dr. John Wojcik, MD, Director, Translational Pathology at Bristol-Myers Squibb from publication: HYPERsol: High-Quality Data from Archival FFPE Tissue for Clinical Proteomics. Journal of Proteome Research. 2020 Jan 14;19(2):973-83.
“For co-IP followed by FLAG peptide elution, we used to do direct in-solution digestion. After one test run with the S-Trap protocol, we completely converted; our results are below. S-Traps work absolutely work with FLAG eluted protein. We are now stocking up on S-Trap micro columns.”
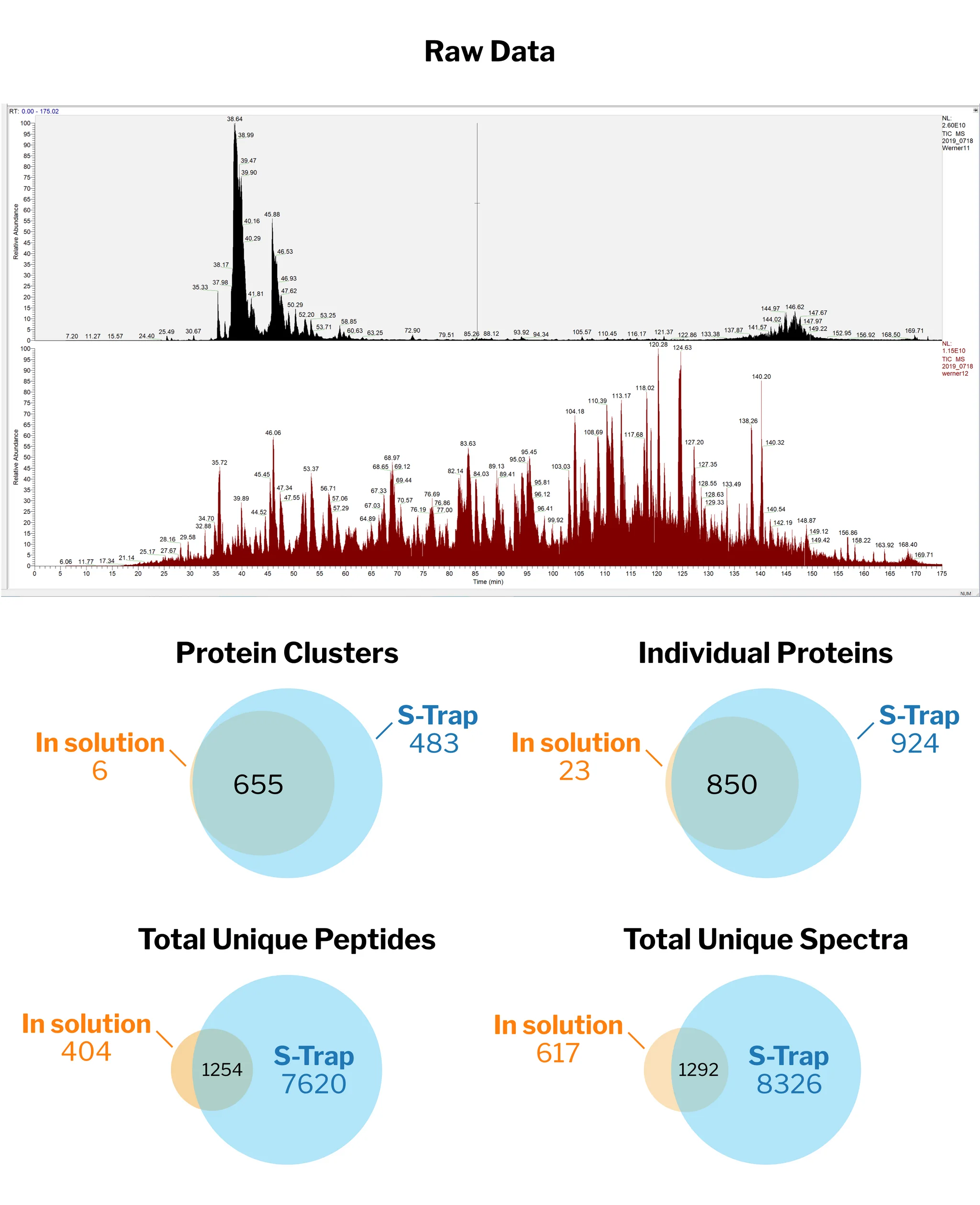
“Incubating mammalian cells in bioreactors requires the addition of polymeric surfactants such as Pluronic F68 [which] are incompatible with mass spectrometry proteomics and must be eliminated during sample preparation. S-Trap substantially reduced or eliminated the polymer(s) and S-Trap provided the most robust cleanup and highest quality data.”